New experimental and computational tools we developed are summarized as follows, including Vicinal, PARIS, SHARC, CRSSANT, dRMS, etc. All of the software and datasets we published can be accessed from Github and NCBI GEO. Additional processed data are available below.
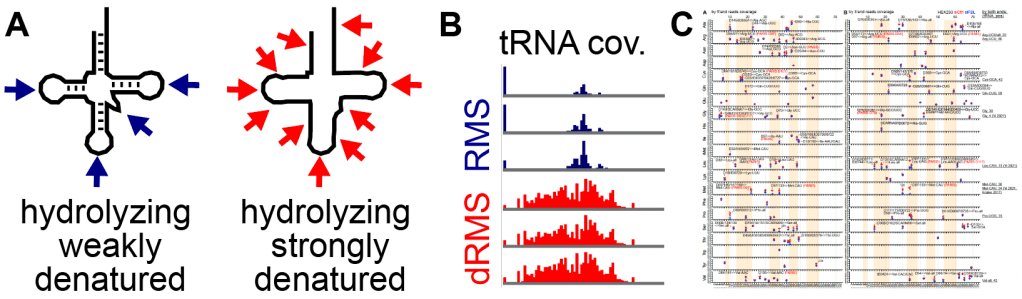
6. dRMS: denatured RiboMeth-seq for 2′-O-me mapping
RNA 2′-O-methylation (Nm) can be mapped based on its ability to block alkaline hydrolysis, however, published methods perform poorly on structured RNAs, especially the shorter ones that are highly modified, such as tRNAs, snRNAs, etc. We developed an optimized dRMS method with superior uniformity and sensitivity, by performing the hydrolysis step under highly denatured conditions (see diagram below). This method reveals more snoRNP-dependent Nm sites among various ncRNAs. Data and detailed protocol is available in Zhang et al. 2023 PNAS. Computational tools are in GitHub.
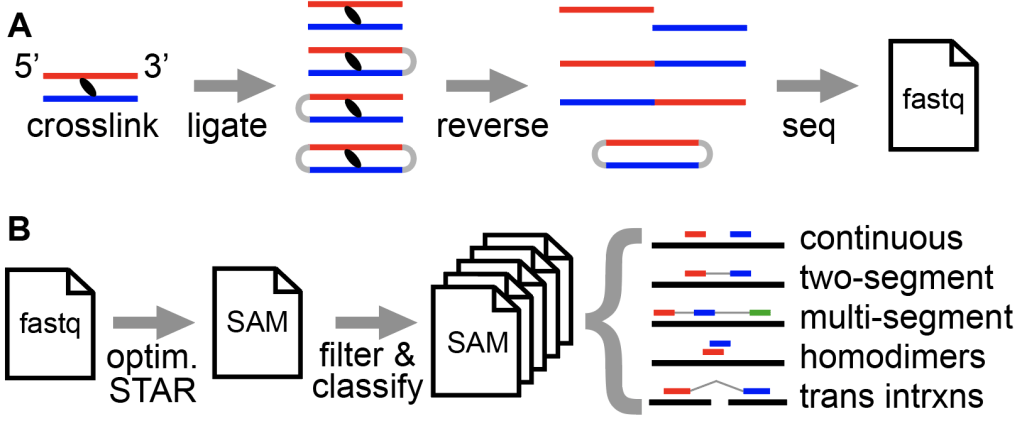
5. CRSSANT: classifying and clustering crosslink-ligation data
RNA crosslink-ligation experiments like PARIS, SHARC, etc. produce gapped reads that can be used to build RNA structure models. CRSSANT (pronounced croissant, following the french theme of PARIS) is a comprehensive package that processes such data, classifies read types, and cluster reads into meaningful groups that support various types of RNA conformations such as complex helix combinations, homodimers, structure dynamics, pseudoknots, triplexes, etc. (Zhang et al. 2022 Genome Research). See GitHub.
4. SHARC: capturing spatial RNA distances to model structures
We developed a new set of chemical crosslinkers called SHARC to measure spatial distances among nucleotides and enable RNA 3D structure modeling in vivo. The new reagents are extremely easy to synthesize: in one step, no purification needed, no special equipment, and very cheap! See publications for details: Van Damme et al. 2022 Nature Communications, Lee et al. Methods Enzymology. For computational methods, see CRSSANT and GitHub. Patent filed.

3. Modular XIST Structures and Functions.
Using a combination of multiple orthogonal computational and experimental approaches, including PARIS, CLIP, fRIP, evolutionary conservation, and genome editing we have built the first whole-transcript-level structure and interaction model for the XIST and several other lncRNP complexes (Lu et al. 2016 Cell. Lu et al. 2020 Nature Comm). Source data for plotting figures in the two papers are available here: XIST_data.
2. PARIS2: Psoralen Analysis of RNA Interactions and Structures
We developed a new strategy to map RNA 2D structurome and interactome in vivo called PARIS (Lu et al. 2016 Cell). We have systematically reinvented the physics, photochemistry, and enzymology in the last 10 years to make it super efficient (PARIS2: >4000 fold better. Zhang et al. 2021 Nature Comm.). Tools for structure analysis and visualization of have been redesigned, see CRSSANT. Original software is available in Github: mine and Cliff’s. Visualization is enabled in IGV, thanks to Jim Robinson and Jill Mesirov.
Raw data from the 2016 paper are available from NCBI (GSE74353). Processed data are provided here from here: HeLa, two replicates, HEK293 and mouse ES, three replicates each (bam and DG bed). RNA interactome data can be downloaded here: HEK293 RNA interactome (Rfam), HEK293 RNA interactome (Rfam + mRNA), mouse ES RNA interactome (Rfam), mouse ES RNA interactome (Rfam + mRNA). The “genome” references can be accessed here: Human Rfam, Human Rfam + mRNA, Mouse Rfam, Mouse Rfam + mRNA

1. Vicinal: chimera analysis for linear RNA ends and circRNAs
Vicinal utilizes locally (which means partially here) mapped reads, from self-priming and ligation, to precisely determine the termini of ncRNAs and provide support for predicated terminal stem-loops (Lu et al. 2014 NAR). Vicinal also uses chimeric reads to discover circular RNAs (Lu et al. 2015 RNA). The scripts, in python and shell, are available from GitHub.
We analyzed hundreds of RNA-seq datasets and compiled lists of ncRNAs with chimeric reads to define their ends, e.g., for snRNAs, scaRNAs, snoRNAs, 7SK, 7SL, RNaseP, RNaseMRP, 5S rRNA, etc. The method does not work for miRNAs, piRNAs, lincRNAs etc. because they do not generate chimeric reads during library preparation. Data are available here: fly_larva3_48nt, fly_ovary_RIP_35nt, fly_pharate_48nt, fly_pupa_48nt, fly_S2_45nt, human_HCT116_50nt, mouse_ES_40nt, mouse_ES_51nt, mouse_satellite_50nt. Each group contains 5 files, including bedgraph, bam and list: prefix_1.bg.gz, prefix_2.bg.gz, prefix_chim_sorted.bam, prefix_chim_sorted.bam.bai and prefix_ncRNA.txt
